If you have hemophilia A (also called classic hemophilia), you are missing or have a deficiency (lower level) of clotting factor VIII (FVIII). This means your blood cannot successfully form a clot. Hemophilia A is hereditary. Because it is an X-chromosome-linked condition, males are more typically affected and therefore more frequently diagnosed. Hemophilia A affects one in 5,000 male births in the U.S. Approximately 400 babies are born with hemophilia A each year. Over 1.1 million people worldwide are living with hemophilia, and about 30,000 are living with it in the United States. All races and economic groups are affected equally.
Most people with hemophilia A who have access to appropriate medical treatment have a normal life expectancy and are able to lead a fairly normal life.
A female carrier of hemophilia who experiences bleeding symptoms is called a symptomatic carrier if her level is over 50%, and she has bleeding symptoms. Carrier females who have between 5% and 50% have mild hemophilia. Hemophilia A severity is the same for women as it is for men. Women with hemophilia and symptomatic carriers have the same symptoms as anyone with hemophilia. Some carriers have completely normal levels of FVIII and do not exhibit any signs of bleeding.
If you have hemophilia A, you should have regular checkups with a hematologist or visit a Hemophilia Treatment Center annually. If you or someone in your family experiences hemophilia A symptoms, and you have not yet been diagnosed, you should contact your medical provider for a referral to a hematologist or HTC for testing and diagnosis.
Hemophilia A is usually hereditary and is an X-linked recessive trait, which means that the defective gene is on the X chromosome. In rare cases, hemophilia A can be acquired due to another medical situation.
Inherited Hemophilia A
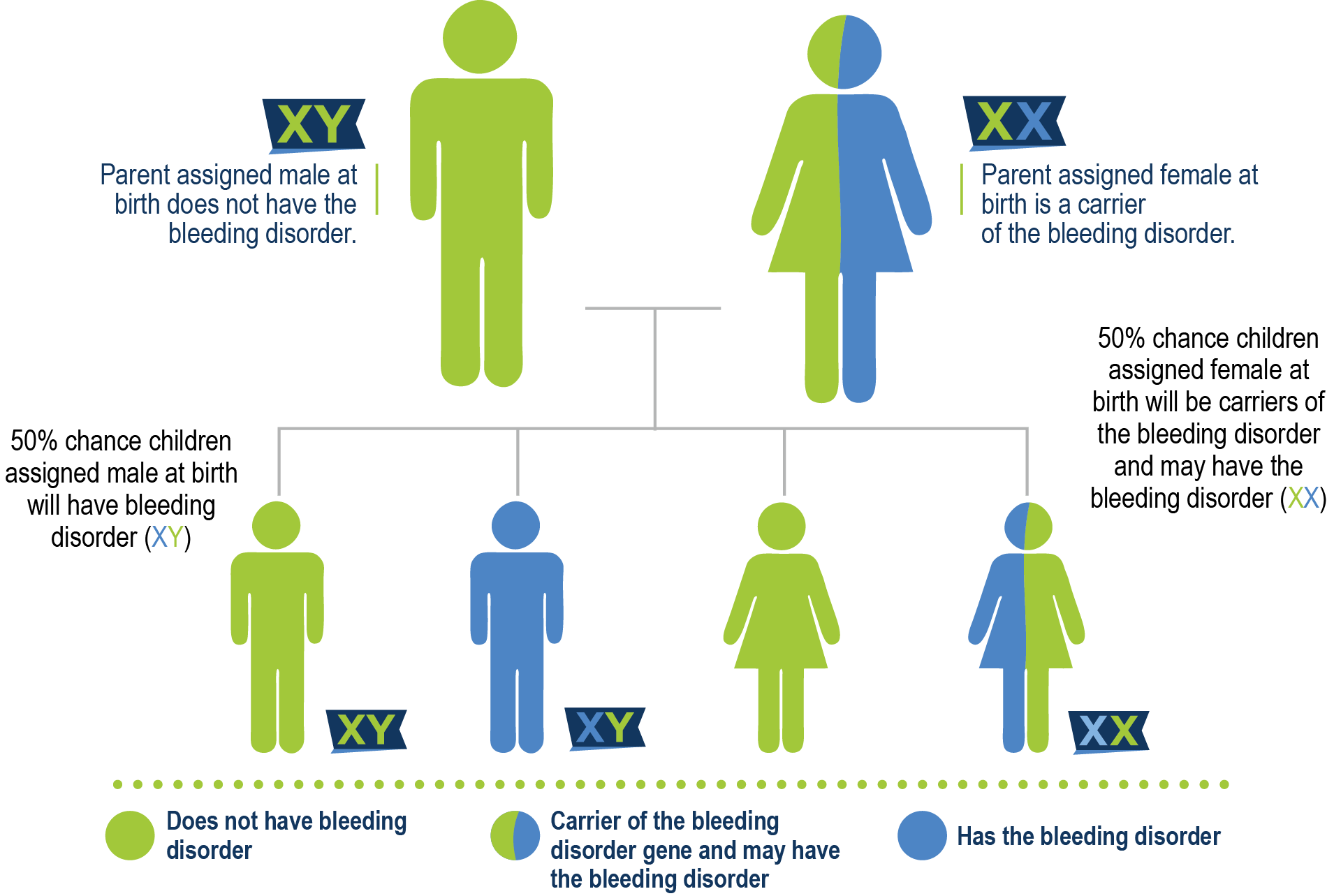
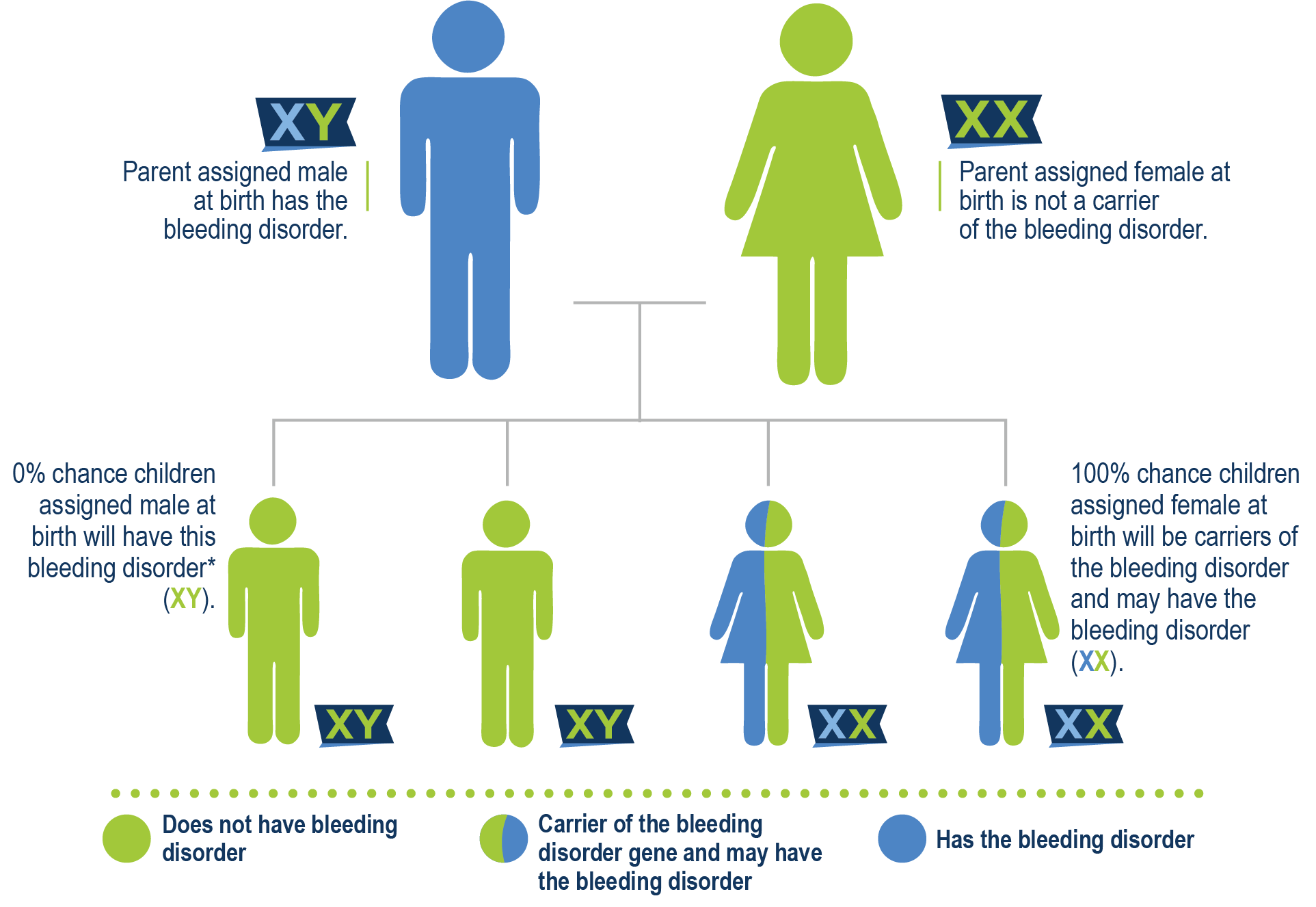
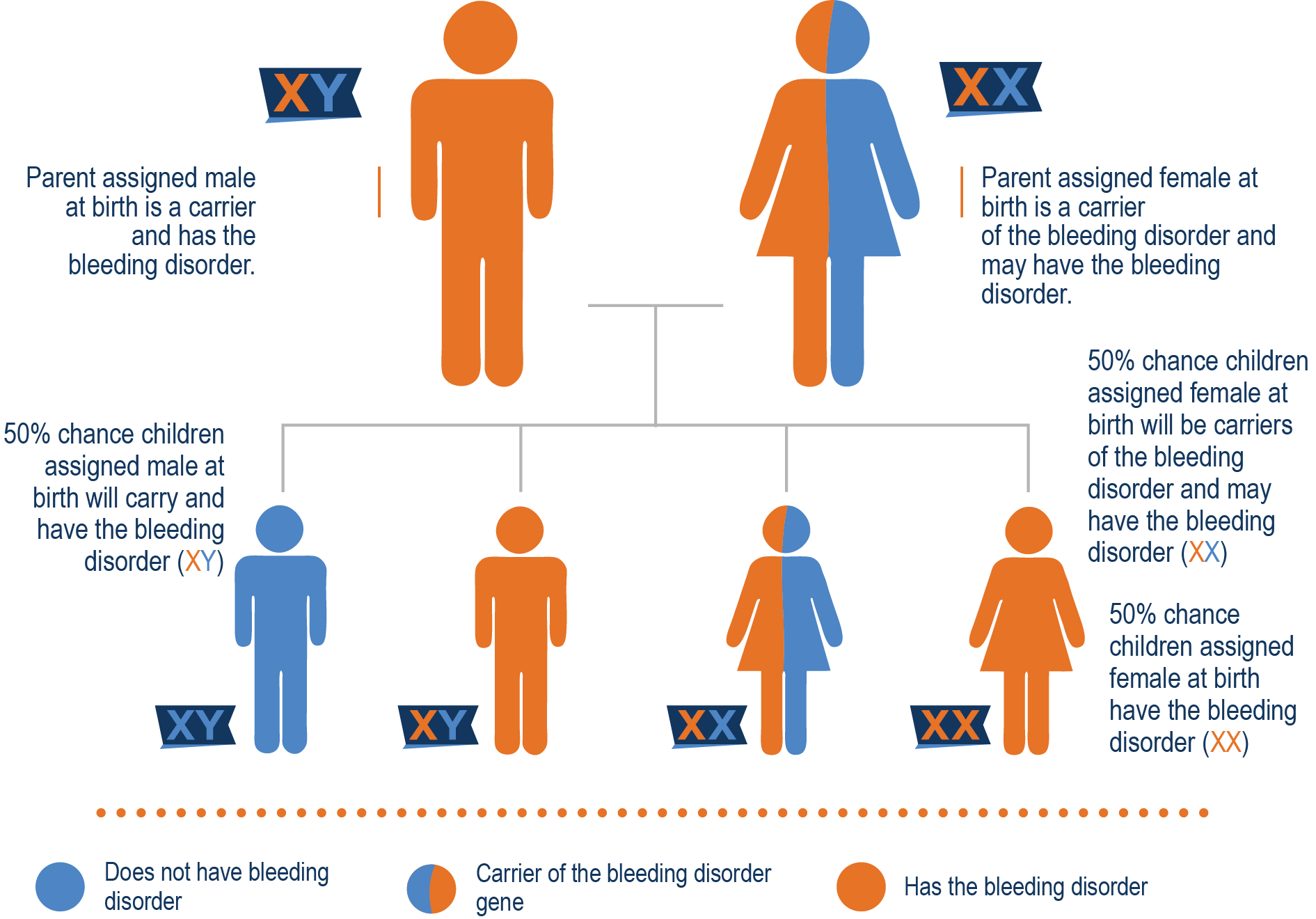
Almost all persons who have hemophilia A are born with it. Hemophilia A is not contagious and does not spread like a virus or an infection. In 70% of the cases of hemophilia A, there is a known family history. The gene that contains the instructions to make factor VIII is on the X chromosome. When that gene is mutated, there can be a lack or deficiency of FVIII. A mother who carries the gene is called a carrier, and she has a 50% chance of having a son with hemophilia and a 50% chance of having a daughter who is also a carrier. A father who has hemophilia possesses the gene and passes it on to his daughter because daughters receive two X chromosomes, one from their mother and one from their father. This is why daughters of men with hemophilia are called obligate carriers. Since sons only receive a Y chromosome from their father, they would not have inherited hemophilia.
Mother is a carrier
Father has hemophilia A
Mother is a carrier and father has hemophilia A
Spontaneous Mutations
Approximately 30% of cases of hemophilia are caused by a spontaneous mutation of the gene. In some cases, the mother is not a carrier of hemophilia, and the child is affected. This child may be the first in the family to have hemophilia and to carry the defective factor gene. In some cases, the genetic mutation occurs in the mother, and she finds out she is a carrier (through DNA testing) after she has a child with hemophilia A.
Female Carriers
Because females have two copies of the X chromosome, if the factor VIII gene on one chromosome does not work, the gene on the other chromosome can sometimes do the job of making enough factor VIII. However, a female might have a genetic FVIII deficiency called “lyonization” or X-inactivation, where some copies of the X chromosome are inactivated. (This is called lyonization because in 1961, a scientist named Mary Lyon proposed the random inactivation of one X chromosome in female mice.) One X chromosome can “shut down” or not express itself. If the functional X chromosome does not express itself, the defective hemophilia A gene takes over and “lyonizes” that functional X chromosome, a female carrier may have lower factor levels.
Acquired Hemophilia A
Hemophilia A may also be acquired, caused by illness, medication or a liver transplant from someone who had undiagnosed hemophilia. This is because clotting factor is made in the liver. Acquiring hemophilia A from a liver transplant is extremely rare.
If you have hemophilia A, you likely bleed longer than someone without hemophilia A, and you can bleed internally or externally. People with hemophilia A do not bleed more than people without hemophilia A; they just bleed longer. The most common sites of bleeding are into the joints and muscles. Baby boys with no family history are often diagnosed when they are circumcised. Other bleeding problems usually show up when babies start crawling and walking. However, if you have mild hemophilia A, you might not be diagnosed until much later in life, possibly after a physical trauma or a surgery.
Common symptoms
- Bleeding into muscles and joints, which can cause pain and swelling
- Nosebleeds
- Prolonged bleeding from cuts, dental work, tooth extraction or surgery
- Easy bruising (unexplained bruising)
- Blood in the urine or stool
- Gastrointestinal tract and urinary tract bleeding
- Bleeding that starts without cause
- Large bruises/hematomas
- Menorrhagia (heavy or prolonged menstrual bleeding: longer than five days, more than 90 ml of bleeding, and/or clots larger than a grape)
Diagnosis
A coagulation study to diagnose hemophilia A might include the following:
- Partial thromboplastin time (PTT)
- Prothrombin time
- Fibrinogen level
- Platelet count
- Serum factor VIII activity
Carrier Testing
A mother who has a son with hemophilia and who has no family history of the condition may or may not be a carrier of hemophilia, depending on where the genetic mutation occurred. A mother who has a son with hemophilia should consider having her DNA tested for her son’s genetic mutation to see if she is a carrier of hemophilia or if the mutation occurred in her son. The genetic information may be important when planning subsequent pregnancies and may be necessary to share with other female family members who are thinking of having children.
Severity
The severity of hemophilia A refers to the amount of clotting factor that is present in a person’s blood. People with severe hemophilia tend to have more frequent bleeding, while people with mild hemophilia may only bleed with a significant trauma, dental procedure or surgery. There are exceptions to this, and not everyone bleeds “by the numbers.”
Factor Level Ranges
- Normal factor levels 50% to 150%
- Mild hemophilia A, 5% to 49%
- Moderate hemophilia A, 1% to 5%
- Severe hemophilia A, less than 1%
Treatment
The treatments for hemophilia A are usually factor replacement therapy, which is administered by infusing (giving medication into a vein) or bispecific antibody therapy, which is administered subcutaneously (a shot right under the skin).
For individuals with mild hemophilia A, desmopressin (DDAVP) might sometimes be used for short-term support. The way it works is that it promotes the release of stored FVIII and von Willebrand factor from endothelial cells in the lining of your blood vessels. It won’t work if you have moderate or severe hemophilia A because you can’t increase something that isn’t there.
In addition to the above products, bleeding may be successfully treated with aminocaproic acid or tranexamic acid. Both of these medications can be given orally.
While anyone with a bleeding disorder can develop an inhibitor to clotting factor, those with hemophilia A develop inhibitors more often than the other bleeding disorders. Inhibitors are antibodies that the immune system develops because it perceives the infused FVIII as a foreign substance that needs to be destroyed. Antibodies are proteins that eat up the FVIII before it has time to stop the bleeding.
Up to 30% of people with hemophilia A are affected by inhibitors at some point in their lives. Inhibitors usually occur between the ninth and 50th infusion of factor concentrate, but in rare cases can also be developed later in life.
While people with severe hemophilia are more likely to develop inhibitors, 5% to 8% of people with mild or moderate hemophilia A also develop inhibitors. In addition to the type and severity of hemophilia someone has, there are other risk factors:
- Age/number of exposures to factor product
- Family history of an inhibitor
- Race/ethnicity (people of African American and Hispanic descent are at a higher risk of developing an inhibitor)
- Gene mutation (there are ongoing studies that indicate the type of genetic mutation one has may indicate a higher risk of inhibitor development)
What are the symptoms of an inhibitor?
Inhibitors destroy the infused FVIII, as well as the FVIII produced naturally by the body in people with mild and moderate hemophilia. If you are treating a bleeding episode, following your current treatment protocol, and the bleeding is not stopping, or may actually be getting worse, this is a key sign that an inhibitor may have developed. Unexplained, more frequent bleeding can also be a sign of an inhibitor. A person who doesn’t have an inhibitor will heal from injuries or feel better after the FVIII is infused or their treatment plan is completed.
How are inhibitors detected?
Anyone with hemophilia A should be tested for inhibitors at least once per year. Testing is a simple blood draw that determines if an inhibitor is present. The inhibitor level is called an inhibitor titer, and that indicates the severity of the inhibitor. The two tests used are called the Nijmegen-Bethesda Assay (NBA) and the Bethesda assay (BA). The NBA titer is measured in Nijmegen-Bethesda units (NBUs), and the BA titer is measured in Bethesda units (BUs), but many doctors will refer to “BUs” regardless of which test is used.
Are all hemophilia A inhibitors the same?
No. Inhibitors come in different degrees of severity. Inhibitors are classified as follows:
Low Titer: When the inhibitor level in the blood is lower than 5 BU. The number, and therefore strength, of the inhibitor is low. People with low-titer inhibitors can sometimes continue to use FVIII products to treat bleeds; they just need a lot more of it. Low-titer inhibitors can sometimes resolve on their own.
High Titer: When the level of inhibitors found in the blood is greater than 5 BU. The number, and therefore strength of the inhibitor, is high. People with a high-titer inhibitor get no benefit from FVIII, no matter how much they infuse.
If you or your child are diagnosed with an inhibitor, your hematologist will likely want you to not infuse for 48 hours to check your BU assay.
Anamnestic Response
An anemnestic response is renewed rapid production of an antibody on the second (or subsequent) encounter with the same antigen. Once an inhibitor is present, the strength with which the body reacts to further exposure of factor concentrate, also called immune response or immunologic response, can further classify the inhibitor type.
Low Responder: When people with low-responding inhibitors receive FVIII, the inhibitor titer does not rise. Because the titer stays low, they may be able to control bleeding by using larger quantities of FVIII concentrates.
High Responder: When people with high-responding inhibitors are exposed to FVIII, the immune system quickly triggers even more inhibitor development. In this case, a bypassing agent may be used to control bleeding.
